The complicated world of splice QTLs
CommentsSummary: There is a lot of interest in analyzing the effect of genetic variants on splicing, but as far as I can tell there is no consensus on the best way to analyze “splice QTLs”. A variety of methods have been used, each answering slightly different questions.
We now have publicly available large scale RNA-sequencing datasets (e.g. from the Geuvadis and GTEx projects, but also from many others) from individuals that also have genotype chip or whole genome sequencing data available. As a result we can now start to identify genetic variants involved in regulating not just gene expression levels, but also in regulating splicing events. There have been a lot of studies in the last several years surveying genetic effects on transcriptomic variation (e.g. Lappalainen et al. 2013, Pickrell et al. 2010, Montgomery et al. 2010, Battle et al. 2013, Zhao et al. 2013, Monlong et al. 2014), but there is no consensus on the best way to analyze “splice QTLs” (sQTLs).
For one thing, while several good tools exist, it is generally agreed that accurately quantifying expression levels of different isoforms is not a solved problem. But another challenge is that unlike gene expression levels, which can be for the most part represented by a single value per gene, there is a confusing variety of ways one might represent the phenotype of an alternatively spliced gene. Each of these representations is best suited to identify differnent types of events, and each comes with its own set of advantages and disadvantages.
So what is the best method to analyze sQTLs? What do most splicing variation events look like? (single exon events? or are they more complex?) How does the sQTL method used affect which events are likely to be picked up? These are some of the questions I have as a relative newcomer to the RNA-seq/splicing world, and from what I can tell we still don’t know the answers.
Below I summarize the methods I’ve seen used in some recent papers and what I think are advantages and disadvantages of each. In my opinion there is no “best method”, and the method you choose is strongly dependent on the type of events you’re looking for or expect to find.
Methods to detect sQTLs
Each method for detecting QTLs has the same general outline: define a phenotype, test for association between that phenotype and the genotype of a nearby variant. For gene-level eQTLs, this is usually straightforward: the phenotype is the expression level of the gene (after some normalization, removing covariates, etc.), and the genotype is something like 0/1/2 for a biallelic variant. For analyzing sQTLs, the question is how do you define the phenotype to capture splicing variation?. Each method below does this slightly differently.
Exon expression level
One widely used phenotype is simply exon expression level. (i.e., for each exon, how many reads mapped to that exon, after normalizing for things like the length of the exon, not going to get into FPKM/RPKM/TPM here).
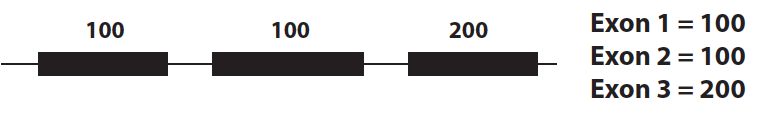
Then you would simply test for association between expression of each exon and each variant nearby. This is one of the methods used by Montgomery et al. and also in the Geuvadis Project paper.
- Advantages: This approach captures variation both at the gene level (classical eQTLs), and variation in splicing level by looking at individual exons separately. The Geuvadis paper finds that many genes have an exon-level QTL but no gene-level QTL, suggesting widespread splicing variability.
- Disadvantages: This method does not give information about transcript structure in particular since each exon is analyzed independently, and so does not allow direct inference of the types of events leading to isoform variability. Also, this method involves doing a lot of non-independent tests, since many exons from the same gene will have correlated expression levels.
- How many are there?: Geuvadis finds 7,825 genes with an exon QTL, compared to only 3,773 genes with a classical gene level eQTL at 5% FDR, suggsting that genetic regulation of splicing variation is more widespread than just variation in expression levels.
Transcript ratio
Another commonly used phenotype is transcript ratio: for each isoform, what percent of the total transcripts from that gene come from that isoform?
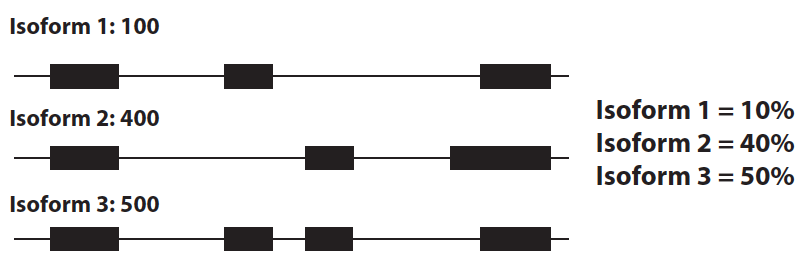
Then you test for association between ratio for each transcript and each variant nearby. This method is used in the Geuvadis Project paper and by Battle et al..
- Advantages: This method captures information about use of different transcripts, and can be used to discern what alternative splicing events tend to look like (skipping a single exon? alternative exon usage? more complex events?). Additionally, unlike exon expression levels, it is specific to splicing events (i.e. it will not pick up gene level eQTLs).
- Disadvantages: It usually relies on known transcript annotations. Therefore, it could be that there is for instance a simple exon skipping event, but if the two different isoforms (one with that exon, one without) aren’t annotated, it would never be picked up. Using known transcript annotations already pre-defines the set of events you can hope to detect. Additionally, it relies on transcript level quantifications which are still not widely trusted. Finally, like the exon expression level method, this method involves many non-independent tests, since transcript ratios for transcripts from the same gene necessarily must sum to 1.
- How many?: Geuvadis identifies 639 genes with “transcript ratio QTLs” at 5% FDR. From the exon level analysis mentioned above, where there were several thousand more exon QTLs than gene QTLs, I would have thought at least several thousand genes show splicing variation. So clearly the transcript ratio method isn’t picking up every event. This could possibly be because: it has reduced power since it’s based on noisier transcript quantifications, or because known transcript annotations don’t capture a large fraction of true splicing events.
Percent exon inclusion (or PSI=percent spliced in)
This method is concerned with inclusion rates of single exons at a time: how often is a given exon included vs. excluded from a transcript?
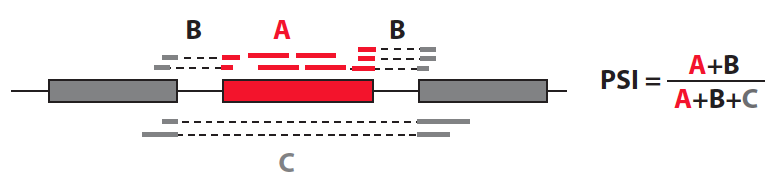
Then you test for association between the PSI of each exon and each nearby variant. Really this is asking: of isoforms that could contain this exon, what percent of them do? This method is used in Monlong et al. to compare to a different method they developed (see below). By design, it specifically captures exon skipping events, but not other types of variation in transcript structure. PSI is mentioned in the Geuvadis Project paper (Figure 4b), although they do not perform QTL analysis on these values. It also forms the basis of the GLIMMPs method by Zhao et al.
A variation on exon inclusion is used in Pickrell et al, where they use as a phenotype the fraction of reads mapped to each exon divided by all reads mapped to that gene. This captures a different set of events than “PSI”: for instance it would capture events involving switching between different transcripts that could differ by multiple exons, whereas PSI is very specific in that it captures only events that differ by exclusion specifically of a single exon.
- Advantages: PSI exclusively captures a specific type of event, simple exon skipping. It also doesn’t have to rely on known transcript annotations, so can capture exon skipping events that wouldn’t be captured using other methods like the transcript ratio method.
- Disadvantages: same as the advantage: it only captures exon skipping events. It wouldn’t be good at picking up switching between two totally different isoforms, alternative 3’ or 5’ ends of exons, etc. Since these complex events seem to be quite abundant, PSI will miss a lot of splice variation events.
- How many?: Monlong et al. find around 400 genes with a PSI QTL per Geuvadis population. Notably, only ~4,500 genes had exons with enough variation in inclusion levels to be tested, indicating that many genes have splicing variations that are not picked up by the PSI score.
Multivariate approach
Monlong et al. take a different approach (called sQTLseekeR) by creating a multivariate phenotype for each gene consisting of the relative abundance of each transcript. (See Figure 1 of their paper).
{kind=link}
They then do a MANOVA like test to find associations between genotype and transcript composition. This is somewhat related to the transcript ratio approach, in that it is testing for variation in which of a known set of isoforms is being expressed, but it involves only one phenotype value per gene, rather than per isoform.
- Advantages: Like transcript ratios, this method captures complex variation in transcript structure. It has the advantage of not having to test each isoform independently as in the univariate approach, which should increase power.
- Disadvantages: It again relies on known transcripts. Therefore, the set of events one can pick up is limited by existing annotations, and also on unreliable transcript quantification.
- How many?: Monlong et al. detect around 180 genes with a sQTL per Geuvadis population using this method (see their table 1). I was surprised that the number was so low, but this could be because they analyze each Geuvadis population separately, and the low sample size could lead to the much lower number of events detected compared to trQTLs from the Geuvadis paper (639). Notably, Monlong et al find only minimal overlap between psiQTLs and sQTLs, suggesting indeed that the methods pick up mostly non-overlapping sets of events.
Conlusion
So what method is best to use? This somewhat depends on what types of splicing events we expect, or what type we hope to find. But what do most splicing events look like? Monlong et al. and the Geuvadis paper used AStalavista to determine the type of events represented by their QTLs. (see Figure 4 of Monlong et al. shown below, plus Figure 2b in the Geuvadis paper).

Both found only ~15% of events to be simple exon skipping, with most events looking more complex (“complex 3’ event”, “complex 5’ event”, alternative exon usage, modified UTRs, etc.). However, it should be noted that these events could be biased by the fact that they used existing transcript annotations for these analyses, which limits the set of events that can be detected. Therefore, this might not represent the true distribution of types of splicing events.
Overall my conclusion is that splicing analysis is complicated! We’re starting to make some progres, but we have a long way to go to make sense of it all.
Tweet comments powered by Disqus